Key word:
Intelligent pharmaceutical design
Rational drug design
3D-RISM Theory
Fundamentals of the research
It's cold season. From sore throats, to high fevers, to runny noses, the symptoms of colds aren't very pleasant. But there is an increasing number of cold medicines commercially available. One might try to choose the best one by oneself by reading the specifications of chemical compounds included in a drug. In fact, one may find a number of different compounds in the specification, acetylsalicylic acid (aspirin), lysozyme chloride, clemastine fumarate, and so on. The specifications say that these compounds are effective for high fever, sore throat, and stuffy nose. However, thinking more carefully about it, how did they know those molecules are really working to ease those symptoms? The size of a molecule, ~10-9 m (one billionth of a meter), is so small compared to the size of human being, ~1 m. Until just few decades ago, it was entirely a puzzle how a drug molecule works in our body to cure a sickness. However, the mechanism of how drug molecules kill a germ such as bacteria or virus has become understood thanks to developments in life sciences including medicine, pharmacology, and biochemistry.
Here, we present research topics concerning "intelligent pharmaceutical design," which has been carried out in Prof. Hirata's group in IMS based on statistical mechanics. Before getting to the topics, we need to become familiar with the meaning of "intelligent pharmaceutical design" in context.
(1) Function of biomolecules, and molecular recognition
In our body, a variety of biomolecules such as proteins and DNA are working hard in order to maintain our life and to pass it on from generation to generation. For example, a protein called "hemoglobin" binds oxygen molecules at lung, and transport them to other organs. It plays a role of "carrier" of oxygen in our body. Therefore, if the protein malfunctions in some reason, we’ll end up sick or even dead. For example, if a human being absorbs too much carbon monoxide, hemoglobin binds the compound preferentially, so that it cannot bind oxygen anymore. That gives rise to so called "carbon-monoxide poisoning."
Proteins play a huge variety of roles, decomposing fat, alcohol, and other compounds (enzymes), adjusting moisture of cells (water channel), and so forth. Such roles played by biomolecules in order to maintain and pass on life are called "biological function." In order for biomolecules to perform their functions, target molecules ("ligand" or "substrate") should bindselectively in some particular place in the biomolecule ("active site" or "reaction pocket"). In the example of hemoglobin, a group called "heme" corresponds to the active site, and oxygen molecule is the ligand. A process in which a biomolecule binds to a ligand is referred to asmolecular recognition. The molecular recognition therefore is the most essential molecular process for a living system. If the molecular recognition process is inhibited for some reason, a human being may get sick or even die.
(2) How does a drug work?
Since germs such as bacteria and virus are also a kind of organism, many proteins and DNA molecules are performing their functions to maintain their lives as well. Whatever the function is, molecular recognition processes are always working there as an elementary molecular process. Therefore, if we can inhibit the molecular recognition processes which are vital for them to maintain their life, the germs will be killed, and we can cure a patient. Many drugs which are prescribed for treating a patient inhibit functions of protein and DNA by binding an active site of those molecules preferentially.
(3) What determines the strength of molecular recognition?
In the preceding paragraph, we mentioned that drug molecules should be bound preferentiallyto an active site of protein or DNA in order to play their role. But, what is the factor to determine the preference? The factor is a quantity referred to as the free energy in terms of thermodynamics. In more plain expression, the quantity is a measure of "discomfort" of a molecule to be in a particular environment: the lower the free energy, more comfortable a molecule. In the case of ligand and drug molecules, if the free energy of a molecule inside the active site is lower compared to that outside of protein or in a bulk environment, then the molecule is bound or recognized by the protein, and it is bound more preferentially as the difference becomes larger and larger. Therefore, it is a crucial step to find a compound of which free energy becomes as low as possible upon the molecular recognition by a target protein or DNA.
So what are the factors that determine the binding free energy? It's easy to imagine using the idea of geometrical compatibility between the geometrical shapes of a drug compound and active-site of protein. This is like the compatibility between lock and key. Another factor which is easy to imagine is the compatibility of the charge distribution in the active site and of the drug molecule. If the electrostatic field in the active site is positive and that of the drug molecule is also positive, then the drug molecule will be repelled out from the active site.
These, however, are not only the factors to determine the free energy. An even greater factor is the environment where protein or DNA is situated, that is, aqueous solutions.Since proteins usually work in an aqueous solution, water molecules are bound at its active-site by default. In order for ligand or drug molecules to be bound to the active site, all or some of the water molecules should be removed. On the other hand, since drug molecules are in aqueous solutions before being bound in the active site, they were already wearing "clothes" of water molecules (we call this hydration). Therefore, a drug molecule, too, needs to undress all or some of its water molecules upon binding the active site of protein.
As is explained above, the binding free energy is affected not only by the geometrical shape and electrostatic charges of protein and a drug molecule, but also the free energy change due to hydration and dehydration.
However, most of the conventional methods practiced in industries and universities in terms of "intelligent pharmaceutical design" have disregarded the free energy of hydration anddehydration. The topics presented here are of a new method of rational drug design, which overcomes the problem of the conventional methods by taking the free energy of hydration anddehydration into consideration.
Research activity
Hirata's group in IMS has been developing a statistical mechanics theory of liquids, referred to as "3D-RISM," and has carried out research concerning structure and functions of biomolecules, which involves molecular recognition. Here, we introduce our recent paper concerning amolecular recognition process of protein, which intends direct application of the 3D-RISM theory to the drug discovery. Our target is a protein called "phospholipase A2 (PLA2)." The protein is known as an enzyme which synthesizes Arachidonic acid, a chemical compound causing pain. Recently, an experiment has disclosed that the compound binds acetylsalicylic acid (or Aspirin) to its active site.(Figure 1) The molecule is regarded as one of model target proteins in the field of drug discovery.
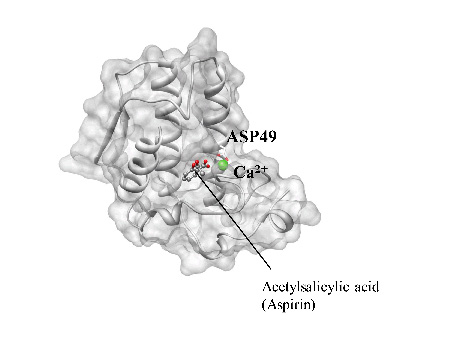
Figure 1. X-ray crystal structure of the binding formed between Phospholipase A2 (PLA2) and acetylsalicylic acid (aspirin). The aromatic ring of aspirin is embedded in the hydrophobic environment and other substituted groups form several important attractive interactions with calcium ion, His48 and Asp49. Calcium ion is shown as a green sphere.
By solving the 3D-RISM equation for a protein-ligand system in aqueous solutions, one can find the three dimensional distribution functions (3D-DF) of ligand around and inside the protein. The 3D-DF is a measure of the probability to find a ligand around a protein. Therefore, the places where 3D-DF take greater value, indicated with red spots in the figure, correspond to those where the ligand binds preferentially. Detailed analyses of 3D-DF naturally provide you information concerning where and how the ligand is bound.
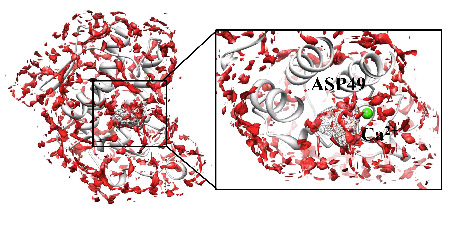 Figure 2. Distribution of acetylsalicylic acid (aspirin), which is obtained by 3D-RISM theory, around and inside Phospholipase A2. The distribution appears the location of acetylsalicylic acid (aspirin) in X-ray structure, which is depicted with a wire frame.
Figure 2. Distribution of acetylsalicylic acid (aspirin), which is obtained by 3D-RISM theory, around and inside Phospholipase A2. The distribution appears the location of acetylsalicylic acid (aspirin) in X-ray structure, which is depicted with a wire frame.
We have defined a score function based on the 3D-DF, and identified the binding mode, or position and orientation, of aspirin around and inside PLA2. The binding mode which scored the top grade is shown in Figure 3. The result shows good agreement with the binding mode deduced from the X-ray crystallography.
Analyses based on a "score function" have already been practiced in the conventional in-silico pharmaceutical design. However, those analyses do not have physicochemical foundation. In other words, the argument is mostly based on the geometrical concept such as "key and lock." The advantage of our method lies on the concept of the three-dimensional distribution functions which have a solid background in statistical mechanics. We believe that the more promising procedure for the drug discovery will become a reality in the pharmaceutical industry once our method of analysis is established.
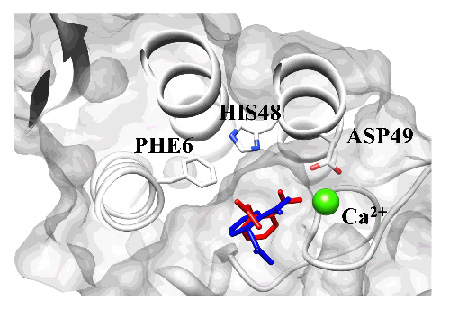 Figure 3. Comparing predicted structure (red) based on 3D-RISM theory with experimental structure (blue) in X-ray analysis. The binding configuration of the ligand inside the pocket was in fair agreement with that determined from X-ray crystallography.
Figure 3. Comparing predicted structure (red) based on 3D-RISM theory with experimental structure (blue) in X-ray analysis. The binding configuration of the ligand inside the pocket was in fair agreement with that determined from X-ray crystallography.
Information of the paper
Journal: Journal of Chemical Theory and Computation (JCTC) 2011, 7(11), 3803-3815. (DOI: 10.1021/ct200358h)
Title of the paper: A New Approach for Investigating the Molecular Recognition of Protein: Toward Structure-Based Drug Design Based on the 3D-RISM Theory
Authors: Yasuomi Kiyota, Norio Yoshida, and Fumio Hirata
Date: September 15, 2011
Group Leader: Fumio Hirata
Professor, Institute for Molecular Science
Home page: http://daisy.ims.ac.jp
