
自然科学研究機構分子科学研究所のJakub Chalupský研究員、倉重佑輝助教、柳井毅准教授、チェコ共和国有機化学生化学研究所のLubomír Rulíšekチームリーダー、Martin Srnec研究員及びスタンフォード大Edward I. Solomon教授による研究チームは、飽和脂肪酸を不飽和脂肪酸へ変換する酵素反応(*2)を高精度量子化学計算を用いて解明することに成功しました。
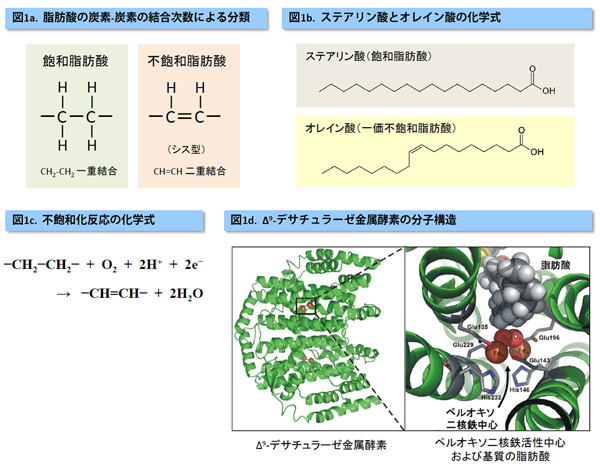
脂肪酸(*1)は炭素が連なった構造を持つ分子ですが、炭素-炭素間に二重結合(C=C)があるかどうかで大きく二つに分類されます。二重結合が無いものは「飽和脂肪酸」、有るものは「不飽和脂肪酸」と呼ばれます。体内では、飽和脂肪酸が不飽和脂肪酸へと変換(不飽和化)するプロセスがあります。その一つは、ステアリン酸(飽和脂肪酸)がオレイン酸(一価不飽和脂肪酸)に変えられる生化学反応です。融点の低いオレイン酸が生成されることにより、体温下で脂肪酸を液状に保ち、流動性を増加させていると考えられています。この変換反応では、「Δ9-デサチュラーゼ」と呼ばれる金属酵素が主たる役割を担うことが知られています。しかしながら、この不飽和化の酵素反応がどのような機構で起こっているかに関する分子レベルでの詳細は未解明でした。
本研究では、分子の様子をコンピュータ上に電子レベルで微細に再現する先端的計算手法を用いて、この反応の律速過程を直接シミュレートし、不飽和化の分子機構を解明することに成功しました。我々の計算結果では、デサチュラーゼ酵素の金属活性中心は、飽和脂肪酸に直接作用するのではなく、タンパク中のプロトン(H+)を活性の助けとして利用して複合的に反応を進めることが明らかにされました。推察された複数の反応機構の中から、この反応機構のみが実験で見積もられた活性化エネルギーと一致することが示されました。このような新陳代謝にも関与する酵素反応の分子論的理解は薬学などでの基礎的知見になることが期待されます。
本研究の実現には、電子の量子的振る舞いを精度よく予測する理論手法の開発が必要でした。本研究チームは、その高精度・高速計算アルゴリズムを開発し、世界で初めて多核金属酵素の反応解析へと応用することができました。この手法は、金属錯体・酵素の触媒作用を高精度にシミュレーションする基盤技術としても期待されます。
本研究成果は、アメリカ化学会の化学雑誌『Journal of the American Chemical Society』オンライン版で公開されました。
研究の背景
金属酵素と呼ばれるタンパク質では、金属イオン(鉄やマンガンなど)を含む化合物が、生命活動に欠かせない生化学反応の活性に深く関わっています。金属酵素は複合高分子であるため、その働きを詳細に理解するには、分光法を用いた実験的解析のほかに、最近では、ミクロな分子の振る舞いをコンピュータ上で直接再現して機構を理解する理論的手法が用いられるようになっています。
金属酵素によって活性される化学プロセスの一つとして、脂肪酸を不飽和化するという酵素反応があります。脂肪酸は、脂質(脂肪や油)の構成要素で、生体のエネルギー源です(図1a)。体内では、余分な糖質やタンパク質から飽和脂肪酸が作られ、さらに不飽和脂肪酸へと変換(不飽和化)されます。この不飽和化反応の一つに、ステアリン酸(飽和脂肪酸)がオレイン酸(一価不飽和脂肪酸)に変えられる酵素反応があります(図1b,1c)。この変換には、「Δ9-デサチュラーゼ」と呼ばれる二つの鉄イオンを補因子として持つ金属酵素が関わっています(図1d)。デサチュラーゼの酵素反応は何段階もの反応ステップを経由して達成されますが、最重要ステップは、飽和脂肪酸の特定のC-C結合部位から水素原子2個を抜き出し二重結合を生成する不飽和化(図1c)です。また、この過程が反応の律速となっていると考えられています。X線結晶解析の結果、不飽和化反応が開始する直前の中間安定構造は特定されていますが、その構造から出発し、どのような分子機構で不飽和化が達成されるかは未解明でした。
このような金属酵素の反応を計算機でシミュレーションするには、「化学結合の糊(のり)」としての役割を持つ電子の振る舞いを精密に計算する必要があります。しかし、その振る舞いを記述する物理の方程式は複雑であるため、従来型アルゴリズムの限界を克服する高速で高精度な計算法(量子化学計算法)の開発が必要とされていました。
研究の成果
本研究では、Δ9-デサチュラーゼ金属酵素によって触媒される脂肪酸の不飽和化の分子機構を高精度量子化学計算から解明しました。今回の研究では、本研究グループが開発した密度行列繰り込み群-多参照二次摂動法(*3)を用いて、これまでにない精度で大規模量子化学計算を実現し、金属酵素の二核鉄イオンの電子状態および不飽和化反応における化学結合の生成と解離の過程をシミュレーションすることに成功しました。
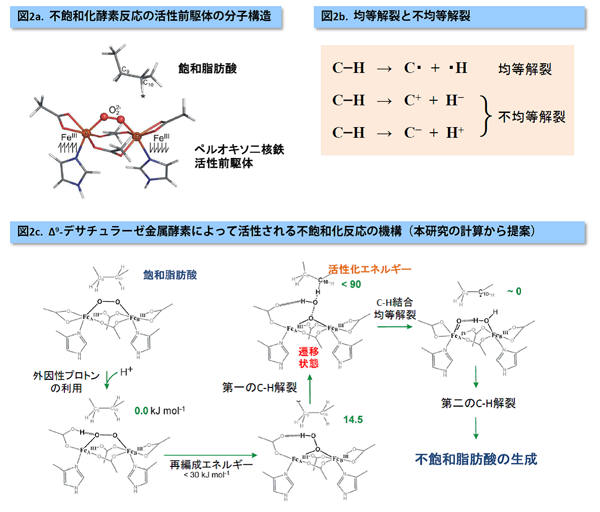
先行の分光実験による研究から、ペルオキソ二核鉄中間体が不飽和化を促す前駆体であることが特定されていました(図2a)。今回の計算では、この前駆体のX線結晶構造を頼りに、そこから出発する不飽和化反応の分子機能の探索を行いました。その際、我々は、反応機構に関して9種類の仮説を立て、それぞれの反応機構の可能性を高精度計算から検証しました。飽和脂肪酸のCH2-CH2部位の両CH2からそれぞれH原子一個を引き抜くことで、一価不飽和酸が生成されるわけですが、本研究の焦点は、最初のHを引き抜くC-H結合の開裂過程の機構解明にありました。この過程は最も活性化障壁が高く重要な対象であり、また、分光実験では解析が難しい反応過程です。我々が立てた仮説としては、
・ペルオキソ二核鉄活性中心が脂肪酸に直接作用し、第一のHを引き抜くモデル
・タンパク中の外因性のプロトン(H+)、水または電子、あるいはそれらの複合を補助因子として利用して反応を進めるモデル
・ペルオキソ二核鉄のO-O結合が先に開裂して活性化するモデル
など化学的に思いつく全ての候補を検証対象としました。
高精度計算に基づく検証の結果、デサチュラーゼ酵素の金属活性中心は、不飽和脂肪酸に直接作用するのではなく、タンパク中のプロトン(H+)を活性の助けとして用いて複合的にC-H開裂反応を進めることが分かりました(図2c)。9種類の反応モデルのそれぞれに関して、活性化エネルギーが高精度計算から得られましたが、本研究が提案するプロトン補助型の反応モデルが唯一実験値を再現するという点が重要な決め手になりました。また、計算された電子の同時確率分布(波動関数)を詳しく解析すると、このC-H開裂は、結合電子2つがCとH原子に均等に移る均等開裂(図2b)とよばれる電子的特徴をもつことが分かりました。この過程では、ペルオキソ二核鉄活性中心のO-O結合も協奏的に開裂します。対照的に、従来型の理論計算(密度汎関数計算)では、C-H開裂を不均等開裂(図2b)として記述し、それが理由で、不飽和化よりもペルオキソのヒドロキシル化を有利な反応として見積もる誤った計算結果を与えます。本研究の高精度計算から予測されたC-Hの均等開裂は、不飽和化を説明する電子論の観点からも理にかなったものと言えます。
今後の展開
本研究で用いられた計算法は、電子レベルの解像度で化学反応をシミュレートできる計算技術であり、これまでにない信頼性を有します。二核金属酵素の反応を高精度に扱った本研究の成果は、本手法の潜在的適応性の高さを示すものであり、その他の金属酵素や金属触媒によって活性される反応系の解析や予測などに幅広く応用されることが期待されます。
また、本研究で明らかにされた不飽和化の分子機構は、分光実験では捉えにくい未解明な反応過程でした。この不飽和化は生体の新陳代謝の一環であり、本研究で示された分子論的理解は、薬学などでの基礎的理解につながることが期待されます。
用語解説
1)脂肪酸:
脂質(脂肪や油)の構成要素で、生体のエネルギー源である。化学的には、長鎖炭化水素の1価のカルボン酸を指す。脂肪の例としてよく知られる中性脂肪は、脂肪酸のグリセリンエステルである。また、脂肪酸とコレステロールの関係は健康問題でしばしば議論される。
2)酵素反応:
酵素は生体の細胞内に存在し、生体内の化学変化に対して触媒・活性作用のあるタンパク質、分子をいう。酵素反応は、酵素によって触媒あるいは活性される生化学反応のことである。
3)密度行列繰り込み群-多参照二次摂動法:
電子の運動を決める量子力学方程式を高精度に効率よく解く計算法の一つ。複数の電子の振る舞いに関する膨大な自由度を繰り込みにより小さな自由度で効率よく表現する密度行列繰り込み群法に基づく方法。
論文情報
掲載誌:Journal of the American Chemical Society(アメリカ化学会誌)
論文タイトル:
“Reactivity of the Binuclear Non-Heme Iron Active Site of Δ9 Desaturase Studied by Large-Scale Multireference Ab Initio Calculations”
(大規模多参照非経験的量子化学計算を用いたΔ9-デサチュラーゼの二核非ヘム鉄活性サイトの反応性に関する研究)
著者:J. Chalupský, T. A. Rokob, Y. Kurashige, T. Yanai, E. I. Solomon, L. Rulíšek, and M. Srnec
掲載日:2014年10月14日 doi: 10.1021/ja506934k
研究グループ
本研究は、自然科学研究機構分子科学研究所・柳井グループ(Jakub Chalupský研究員、倉重佑輝助教、柳井毅准教授)、チェコ共和国有機化学生化学研究所(Lubomír Rulíšekチームリーダー、Martin Srnec研究員、Tibor András Rokob研究員)及びスタンフォード大Edward I. Solomon教授との共同研究として行われました。
研究サポート
本研究は、科学研究費補助金の基盤研究(B) 25288013、基盤研究(C) 25410030、新学術領域研究「柔らかな分子系」26104538の一環として行われました。
研究に関するお問い合わせ先
柳井 毅
理論計算分子科学研究領域、准教授
TEL:0564-55-7301
E-mail:yanait_at_ims.ac.jp
報道担当
自然科学研究機構・分子科学研究所・広報室
TEL/FAX 0564-55-7262
E-mail: kouhou_at_ims.ac.jp
1010